[ブログvol.2] 赤外スペクトルの構造、読み方(解析)、代表的なピークやスペクトル — 基礎編
赤外スペクトルの基本構造
赤外スペクトルは通常、横軸に波数(cm⁻¹)、縦軸に透過率(%)または吸光度(Abs)をとって描かれます。波数は値が大きいほど高エネルギー(短波長)の振動を意味し、赤外スペクトル上では一般的に左側が高波数、右側に低波数で表示されます。典型的な中赤外の測定範囲は4,000 cm⁻¹から400 cm⁻¹程度で、1,500 cm⁻¹以下は「指紋領域」と呼ばれ、多くの分子構造情報を含みます。スペクトル上のピーク(吸収ピーク)は試料分子中の特定の化学結合の振動に対応しています。例えば、O-H結合の伸縮振動は3,600–3,200 cm⁻¹付近、C=O結合の伸縮振動は1,850–1,630 cm⁻¹付近に現れることが知られています。各ピークの位置(波数)、強度、形状からその由来となる官能基を推定できます。
代表的なピークの例
以下に赤外スペクトルでよく確認されるピークと対応官能基の例を一部紹介します。
O-H伸縮(ヒドロキシ基): 約3,200–3,600 cm⁻¹に幅広い吸収帯として現れます。アルコールのOHは約3,300 cm⁻¹付近で丸みを帯びた広いピークになり、水素結合の程度によって幅や位置が変わります。カルボン酸のOHはさらに低波数側(3,000 cm⁻¹付近)に非常に広いピークを示します。
C-H伸縮: 3,100 cm⁻¹付近以上に現れるピークは=C-H(アルケンや芳香環のC-H)に対応し、3,000 cm⁻¹以下(2,800–3,000 cm⁻¹)のピークは-C-H(アルカンのC-H)に対応します。例えばメチレン基(–CH₂–)の不対称伸縮は約2,922 cm⁻¹、対称伸縮は約2,850 cm⁻¹に出現し、メチル基(–CH₃)の対応ピーク(約2,956 cm⁻¹, 2,875 cm⁻¹)とあわせて複数の峰が確認できます。
C=O伸縮(カルボニル基): 約1,800–1,650 cm⁻¹に強いピークが現れます。具体的にはケトンやカルボン酸では1,710–1,730 cm⁻¹、エステルでは1,730–1,750 cm⁻¹、アミドでは1,650 cm⁻¹付近など、官能基によって位置が異なります。
C=C伸縮(炭素二重結合): 非共役のアルケンのC=Cはおよそ1,640–1,680 cm⁻¹に中程度の強さで現れます。芳香環のC=C振動は1,600 cm⁻¹付近と1,500 cm⁻¹付近に2本現れることが多いです。例えばベンゼン環では~1,600 cm⁻¹と~1,500 cm⁻¹にピークが現れ、特に1,600 cm⁻¹付近のピークはポリスチレンの検量線にも用いられる基準ピークです。
指紋領域の吸収: 1,500–500 cm⁻¹付近は「指紋領域」と呼ばれ、C–O伸縮、C–N伸縮、C–Cl伸縮、CH₂やCH₃の変角振動(曲げ振動)など多くのモードが密集します。例えばエステルのC–O伸縮は1,200–1,100 cm⁻¹に強いバンド群として現れますし、メチレン基は1,300–700 cm⁻¹付近に現れます。指紋領域は分子ごとにパターンが大きく異なるため、化合物の同定(照合)に利用されます。
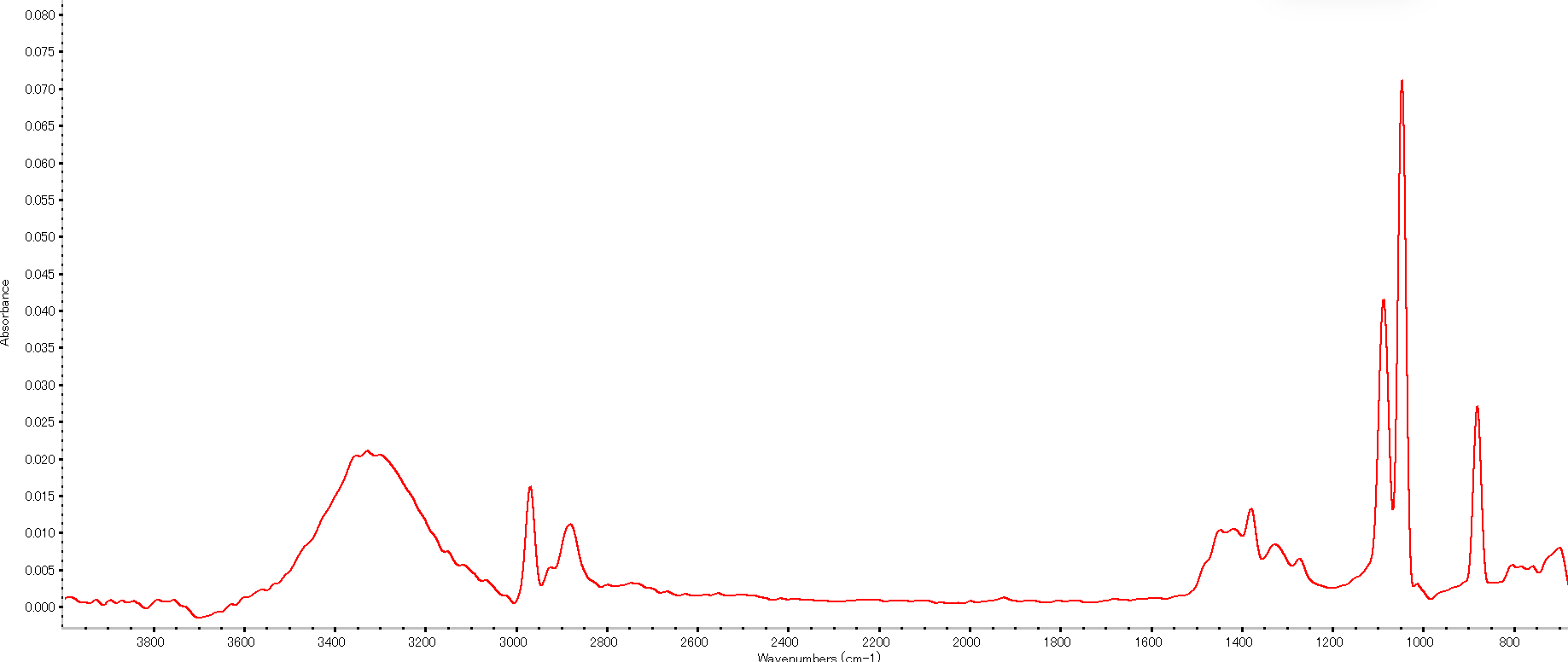
上図はエタノール(CH₃CH₂OH)の赤外透過スペクトルの例です。波数軸は右端が600 cm⁻¹、左端が4,000 cm⁻¹で、縦軸は吸光度(Abs)です。3358 cm⁻¹付近に幅広い山が見られますが、これはOH基の伸縮振動に由来する吸収です。アルコールのOHピークはこのように非常に幅広い形状を示し、水素結合により中心波数がやや低下し幅が広がります。一方、2974 cm⁻¹や2887 cm⁻¹付近の比較的鋭いピークはC–H伸縮振動に対応します。エタノールにはメチル(–CH₃)とメチレン(–CH₂–)が含まれ、それぞれ不対称・対称振動で複数のピークを与えるため、約2,900 cm⁻¹付近に密集したピーク群が見えています。1455 cm⁻¹と1381 cm⁻¹付近のピークはCH₂・CH₃の変角振動に由来します。1090 cm⁻¹と1050 cm⁻¹付近のピークはC–O伸縮振動です。エタノール中のエーテル結合(C–O–H)がこの領域に特徴的な2本のピークを与えています。880–670 cm⁻¹付近には小さなピークがいくつか見えていますが、これは主にC–C骨格振動やC–Oの変角振動などに対応する指紋領域の吸収で、669 cm⁻¹のピークはエタノールに固有の吸収の一つです。
赤外スペクトルの読み方(解析の基本)
赤外スペクトルを解析する際、一般的には、測定スペクトルとライブラリスペクトルの相関により、自動で候補物質を提示してくれますが、スペクトルを読み取るためには、まず2,3か所の特徴を見つけるのがコツと言われます。例えば、一つは3,200–3,600 cm⁻¹付近の広いピーク(OHやNHの有無)、もう一つは1,600–1,800 cm⁻¹付近のピーク(C=Oの有無)です。最初にこの領域でブロードなOHピークやC=Oのピークが存在するかを確認します。もし未知試料の赤外スペクトルに幅広いOHピークがあればアルコールもしくは酸だと推定され、強いC=Oピークがあればカルボニル化合物だと分かります。次に2,800–3,000 cm⁻¹のC–Hピークの数や位置から、試料が脂肪族か芳香族か、メチル基を持つか等を推定します。例えばピークが4本あればCH₃とCH₂両方含む、2本だけならどちらか一方だけ、という判断が可能です。その後、1,000–1,500 cm⁻¹の指紋領域パターンを参考に、候補化合物のスペクトルデータベースと照合します。指紋領域は複雑ですが、その分スペクトル照合による化合物同定には有効です。最後にピークの強度比から定性的な含有量を推察したり、水やCO₂による妨害をチェックしたりします。例えばスペクトル中に1,640 cm⁻¹や3,500 cm⁻¹付近の小さいピーク群やノイズ群が見える場合、それは大気中の水分の吸収によるものかもしれません。
このように、赤外スペクトル解析は「まず大まかな特徴を掴み、次に細部を照合する」という手順で行うのが効率的と言われます。重要なのは優先順位をつけてスペクトルを眺めることだと思います。とはいえ、赤外スペクトルは一般的に混合物のスペクトルであることがほとんどであるため、FTIRメーカーから供給されている多成分検索ツールを利用したり、或いは測定手法やサンプリング方法、差スペクトル処理など工夫しながら、混合物や不純物を見極めることが必要です。
オススメの教材
高山森先生による実用的な赤外スペクトル集を紹介いたします。『赤外スペクトルによるプラスチック・エラストマーの定性 Vol. 基礎編 赤外スペクトルの読み方』は、「赤外スペクトルの解析は難しい」と感じているすべての技術者・研究者にとって、頼れる一冊です。詳細は下記リンクよりご確認ください。
https://drl.official.ec/items/124626050
